![]()
对“分子动力学”的认识最初来自于本科时的《计算材料学》课程。那时候就听说有个叫做LAMMPS的开源软件,可以实现分子动力学模拟。但是苦于安装困难,并没有继续深究。作为一个热爱折腾的人,没有深究这件事一直让我过意不去。
好在,时过境迁。今天的LAMMPS早已不是过去的LAMMPS,行云流水的安装进度让人止不住地鼓掌叫好。那么,究竟有多简单?
Windows 10系统上安装
在Windows上的安装,可以直接访问美国Sandia国家实验室地官网:https://lammps.sandia.gov/。进入Download界面,找到你所需要的Windows版本。说时候,网站导航不是一般的迷惑,很容易迷路,所以在这我给大家一个参考:关键词是Pre-built Windows executables,含义是已经编译好的Windows可运行程序(exe文件)。如果你是开发者的话,也可以选源代码,自己编译代码运行,灵活度更高。
下载下来一步步安装就好了。然后我们会收获一个文件夹。
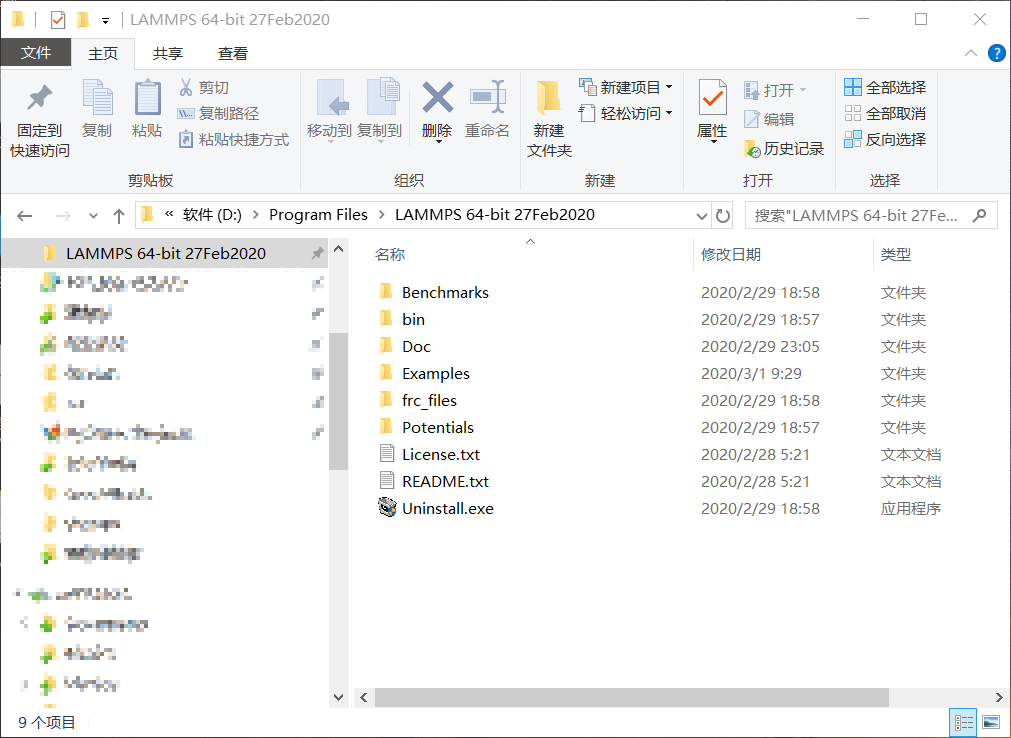
好了,你没看错,我们什么都不需要配置,已经获得了一个可以输出有趣结果的LAMMPS程序了!需要在Linux编译,还有很多奇怪的参数需要配置的日子已经一去不复返了。
运行LAMMPS
正确的文件夹
我们可以跑个demo(示例)试试。记住以下路径:
1 | 当前目录(即LAMMPS根目录)/Examples/colloid |
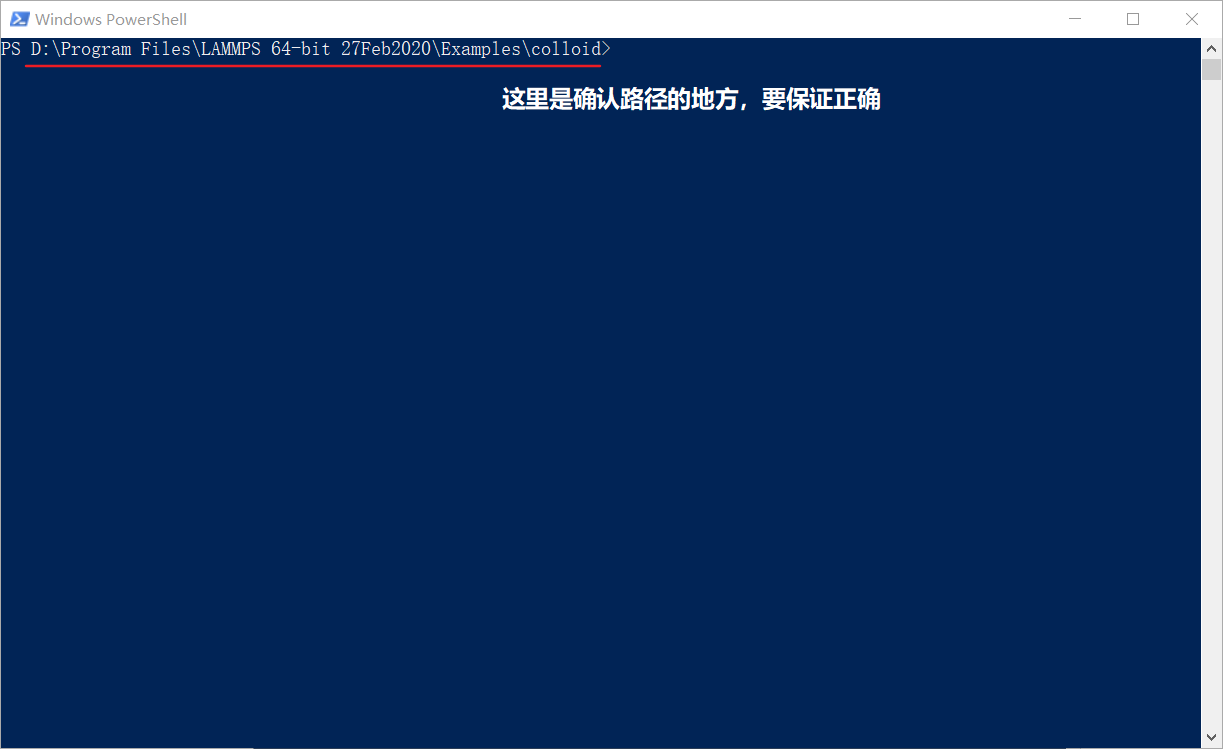
这就是一个简单的colloid示例所在的项目位置。LAMMPS启动的方式很GEEK:先进入以上文件夹(colloid),右键单击后,选择在此处打开Powershell(cmd也可以,俗称小黑框):
输入ls,可以显示当前文件下下的文件:
1 | PS D:\Program Files\LAMMPS 64-bit 27Feb2020\Examples\colloid> ls |
或者输入dir(cmd中没有ls命令):
1 | PS D:\Program Files\LAMMPS 64-bit 27Feb2020\Examples\colloid> dir |
可以看到文件夹里有一个叫做in.collide的文件。这便是LAMMPS的输入文件了。更改输入文件,我们可以实现不同模型的计算。对原子/分子结构和坐标还可以用data文件定义,我们之后再进行讨论。
一定要进入有in.collide文件的文件夹,要不然下一步操作是无效的。
正确的运行
接下来输入一个简单的命令lmp_serial -i in.collid(这是最重要的):
1 | PS D:\Program Files\LAMMPS 64-bit 27Feb2020\Examples\colloid> lmp_serial -i in.collid |
如果出现下列状态,恭喜你,运行成功了。
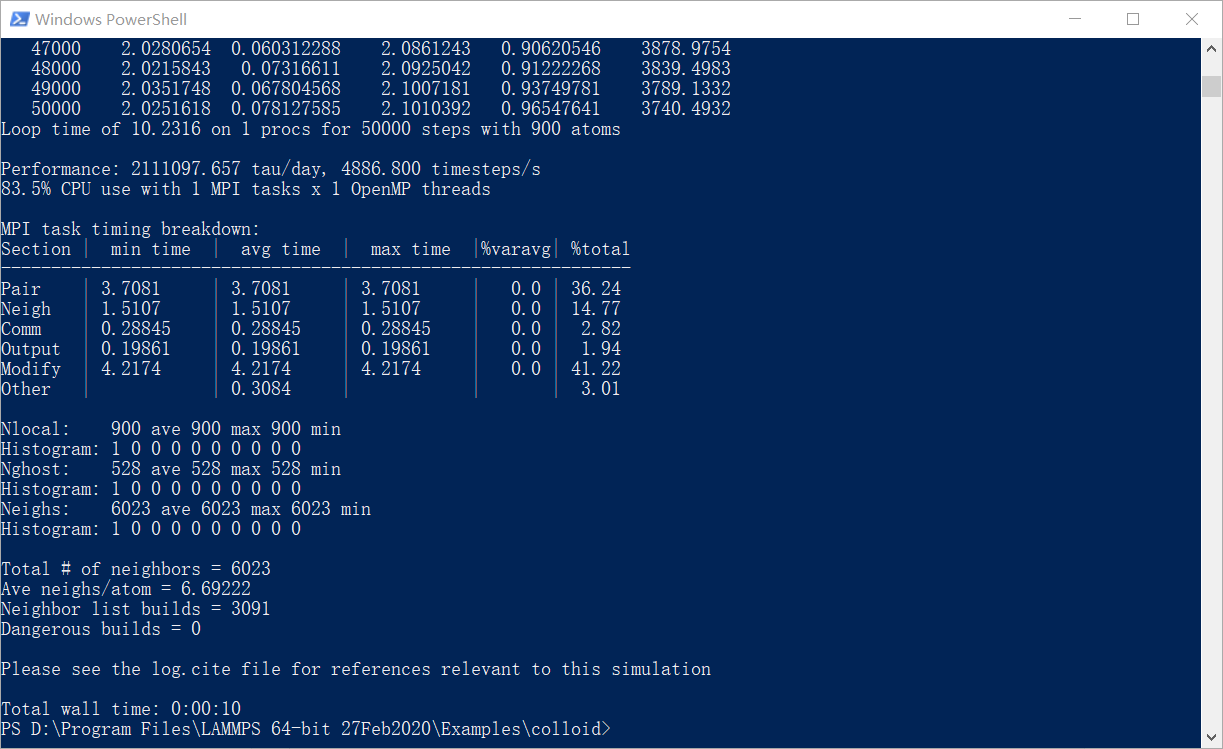
知其然,还要知其所以然。这命令是什么意思呢?
我试着查了一下,输入lmp_serial -h(-h参数常做help使用):
1 | PS D:\Program Files\LAMMPS 64-bit 27Feb2020\Examples\colloid> lmp_serial -h |
OK,我们这里的lmp_serial是串行(serial)的版本的LAMMPS;-i的意思是”read input from file“,即从in文件中读取关于LAMMPS运行的一切模型和参数;最后接上in文件的名字in.collid,就大功告成了。
对应串行运行命令
lmp_serial的是并行运行命令lmp_mpi。如果在计算中心进行模拟,多是并行多核计算。
写在后面
博主发现LAMMPS这个软件本身很强大好用,可用于较多现象的模拟,但在国内的应用却并不多,相关资料更是极其缺乏(即使是收费资料)。博主自己琢磨的过程走了很多跟科学无关的弯路,在这里记录下来,希望大家使用的时候都能少走弯路。
到此为止我们已经能够让LAMMPS正常运行了。下次我们的主题将会是如何掌握程序的输入输出,实现脚本调试。而怎么让LAMMPS根据物理规律和我们的意图正确运行,将是我们下下次的讨论的主题。Love and peace。